PoET: A High-Performing Protein Language Model for Zero-Shot Prediction
PoET, our new generative protein language model, can controllably generate protein sequences de novo, and make zero-shot variant effect predictions for protein engineering and clinical variant prioritization with state-of-the-art accuracy. This blog post explores PoET's capabilities in terms of speed, accuracy, generalizability, and ability to generate context-relevant predictions. PoET is available through our web app and APIs at OpenProtein.AI. More information about the model can be found in our docs, our publication, and our code on GitHub.
Protein language models are poised to revolutionize protein engineering. By leveraging deep learning to uncover complex relationships between sequence, structure, and function, protein language models drive unprecedented accuracy and efficiency in protein design and optimization. Just as natural language processing (NLP) models, like ChatGPT, learn from large text datasets to understand human language, protein language models are trained on extensive natural protein sequence databases. While NLP models predict word probabilities to create coherent text, protein language models predict amino acid probabilities in natural sequences, learning the statistical constraints that govern functional and stable proteins.
Current protein language models, while powerful, have several limitations that hinder their practical utility. Increasing model size yields diminishing returns in accuracy at the cost of slower, more expensive inference. Current models fail to effectively handle insertions, deletions, and generalization to novel protein families. They also lack user-directed control through constraints or objectives. These limitations restrict the real-world applicability of these models.
Introducing PoET: The Protein Evolutionary Transformer
To address the challenges biologists face when using ML for protein engineering or variant effect prediction, we developed PoET (the Protein Evolutionary Transformer). PoET is a generative protein language model that employs unsupervised machine learning to model evolution across protein families, making predictions about protein sequence fitness and generating novel proteins. Similar to how ChatGPT uses prompt-tuning for controlled text generation, PoET is homology-augmented, utilizing contextually relevant user-provided prompts to focus on specific local sequence spaces, from antibodies to enzymes to structural proteins. By training on hundreds of millions of sequences spanning diverse protein families, PoET learns both common and family-specific evolutionary patterns. During prediction, it uses user-provided prompts to efficiently adapt to new protein families and contexts without the need for costly retraining.
Unlike existing methods that rely on multiple sequence alignments (MSAs), PoET models protein families directly as sets of sequences, learning to align sequences itself and avoiding issues caused by long insertions, gaps, and alignment errors. PoET's novel architecture enables efficient modeling of large context lengths (i.e., more sequences), resulting in an accurate, fast, and scalable model for controllable zero-shot predictions and de novo sequence generation.
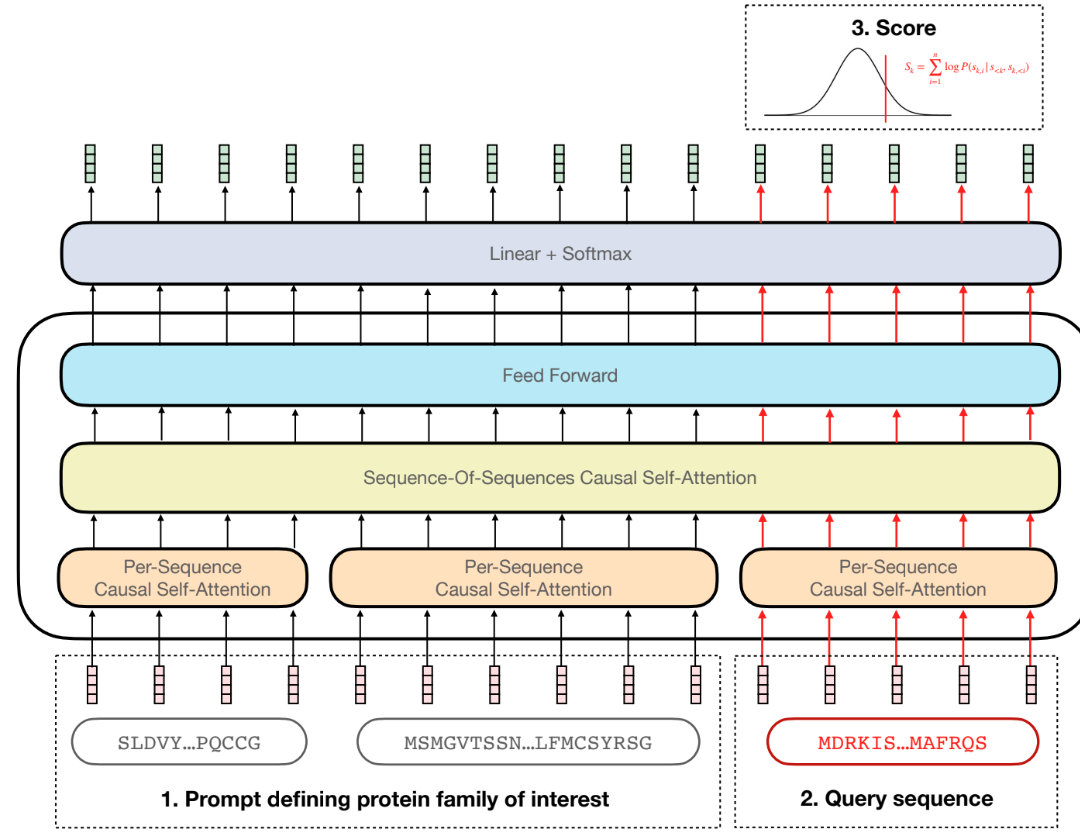
Zero-Shot Prediction Capabilities
PoET's zero-shot prediction capability enables it to predict the effects of mutations in protein sequences, helping biologists understand the impact of substitutions, insertions, deletions, and higher-order mutations before collecting experimental data. The model reports predicted sequence fitness as an evolutionary log-likelihood value, reflecting properties crucial for a protein's natural function, such as stability, catalytic efficiency, and binding ability. This emerges, in part, from the strong relationship between protein stability and fitness (see Echave & Wilke, 2017 for more on biophysical models of protein evolution).
Performance Benchmarks
We benchmarked PoET's zero-shot prediction performance using Deep Mutational Scanning (DMS) datasets from ProteinGym, a comprehensive collection of experimental datasets spanning different taxonomies, functions and properties, and types of mutations. DMS datasets quantitatively measure protein variant performance across various assays, including binding, aggregation, and thermostability. Comparing PoET log-likelihood scores to measured values using Spearman correlation, we demonstrated PoET's ability to accurately predict functional protein variants.
PoET outperforms existing models on both substitution and indel variants (unlike PoET, models like ESM cannot predict insertion and deletion effects). Despite relying on evolutionary data for conditioning, PoET excels on proteins regardless of natural homolog count (MSA depth). It also performs well across assay types, taxonomic groups, and when predicting higher-order mutation effects.
Substitutions By MSA Depth
| Model | Low | Medium | High | All | Indels |
|---|---|---|---|---|---|
| PoET | 0.488 | 0.476 | 0.518 | 0.474 | 0.519 |
| ESM-1b | 0.350 | 0.398 | 0.482 | 0.394 | N/A |
| ESM-1v | 0.326 | 0.418 | 0.502 | 0.410 | N/A |
| ESM-2 | 0.335 | 0.406 | 0.515 | 0.414 | N/A |
| MSA Transformer | 0.404 | 0.450 | 0.488 | 0.434 | N/A |
| ProGen2 | 0.354 | 0.405 | 0.444 | 0.391 | 0.434 |
| TranceptEVE | 0.451 | 0.467 | 0.492 | 0.456 | 0.416 |
| GEMME | 0.454 | 0.470 | 0.497 | 0.456 | N/A |
Performance by Assay Type
| Model | Activity | Binding | Expression | Fitness | Stability | All |
|---|---|---|---|---|---|---|
| PoET | 0.500 | 0.401 | 0.467 | 0.481 | 0.519 | 0.474 |
| TranceptEVE | 0.487 | 0.376 | 0.457 | 0.460 | 0.500 | 0.456 |
Performance by Taxonomic Group
| Model | Human | Other Eukaryote | Prokaryote | Virus | All |
|---|---|---|---|---|---|
| PoET | 0.483 | 0.536 | 0.475 | 0.497 | 0.474 |
| TranceptEVE | 0.471 | 0.498 | 0.473 | 0.453 | 0.456 |
Performance by Number of Mutations
| Model | 1 | 2 | 3 | 4 | 5+ | All |
|---|---|---|---|---|---|---|
| PoET | 0.471 | 0.294 | 0.431 | 0.392 | 0.440 | 0.474 |
| TranceptEVE | 0.446 | 0.277 | 0.350 | 0.320 | 0.382 | 0.456 |
Spearman correlation values of PoET and other commonly used open-source protein language models across different categories within ProteinGym's Deep Mutational Scanning dataset.
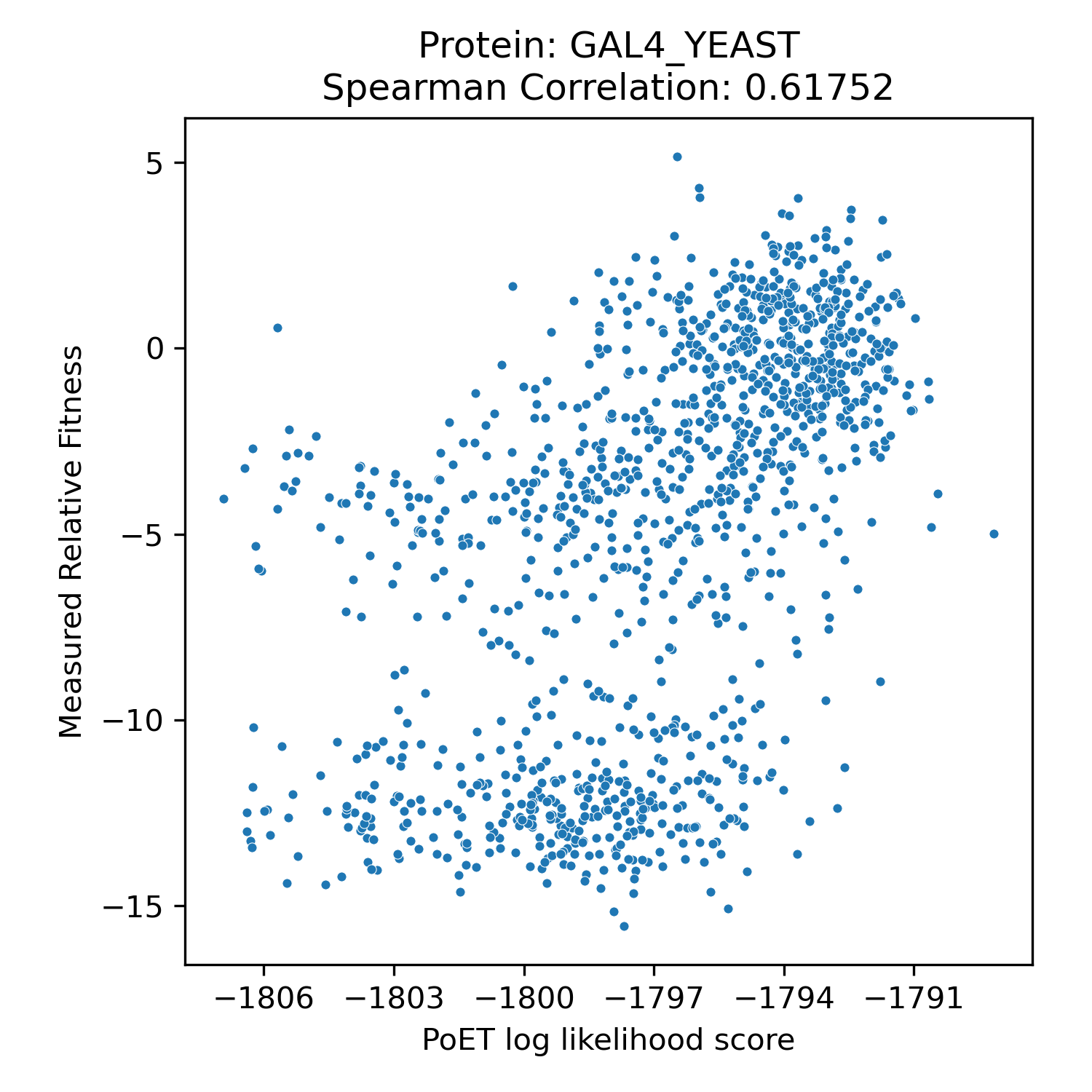
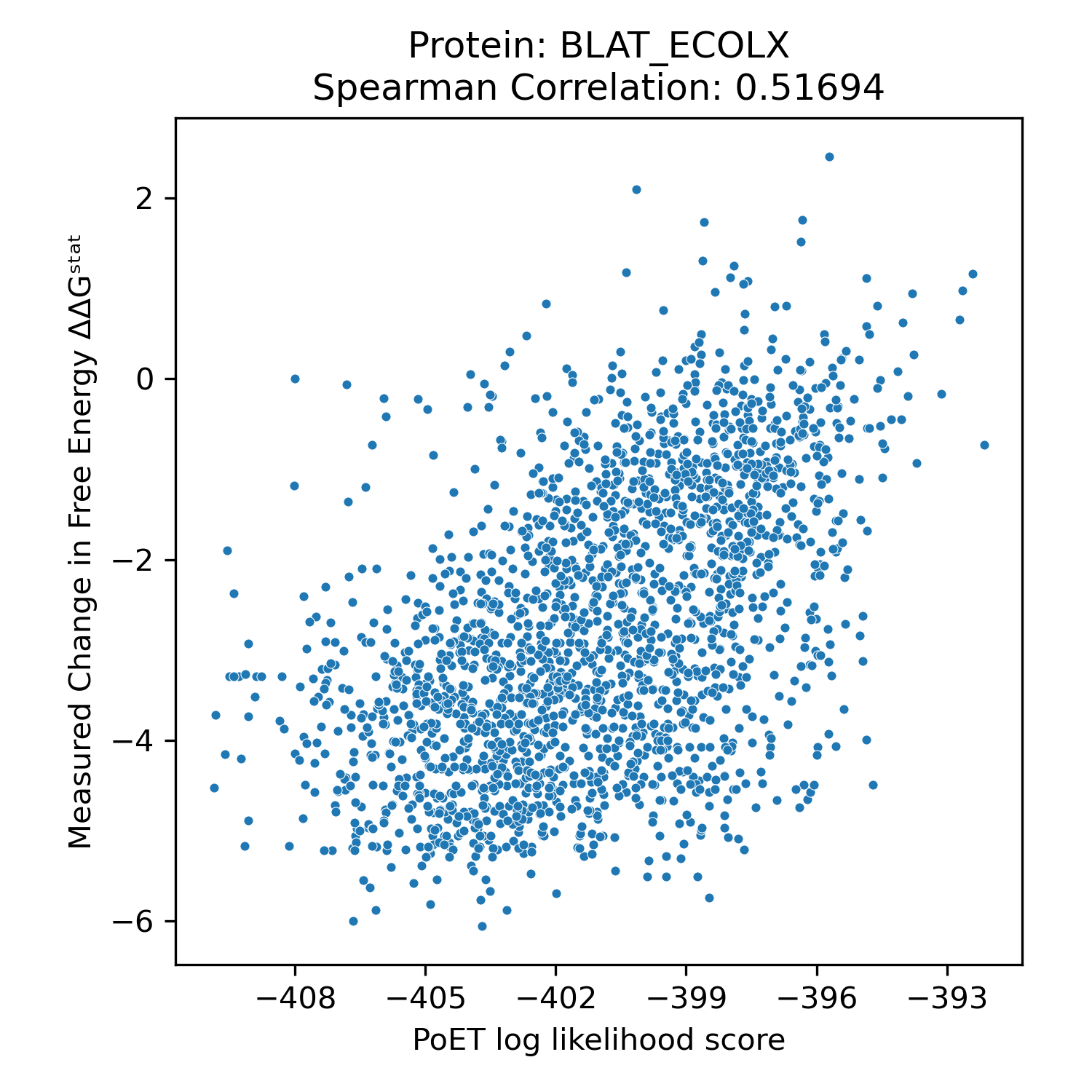
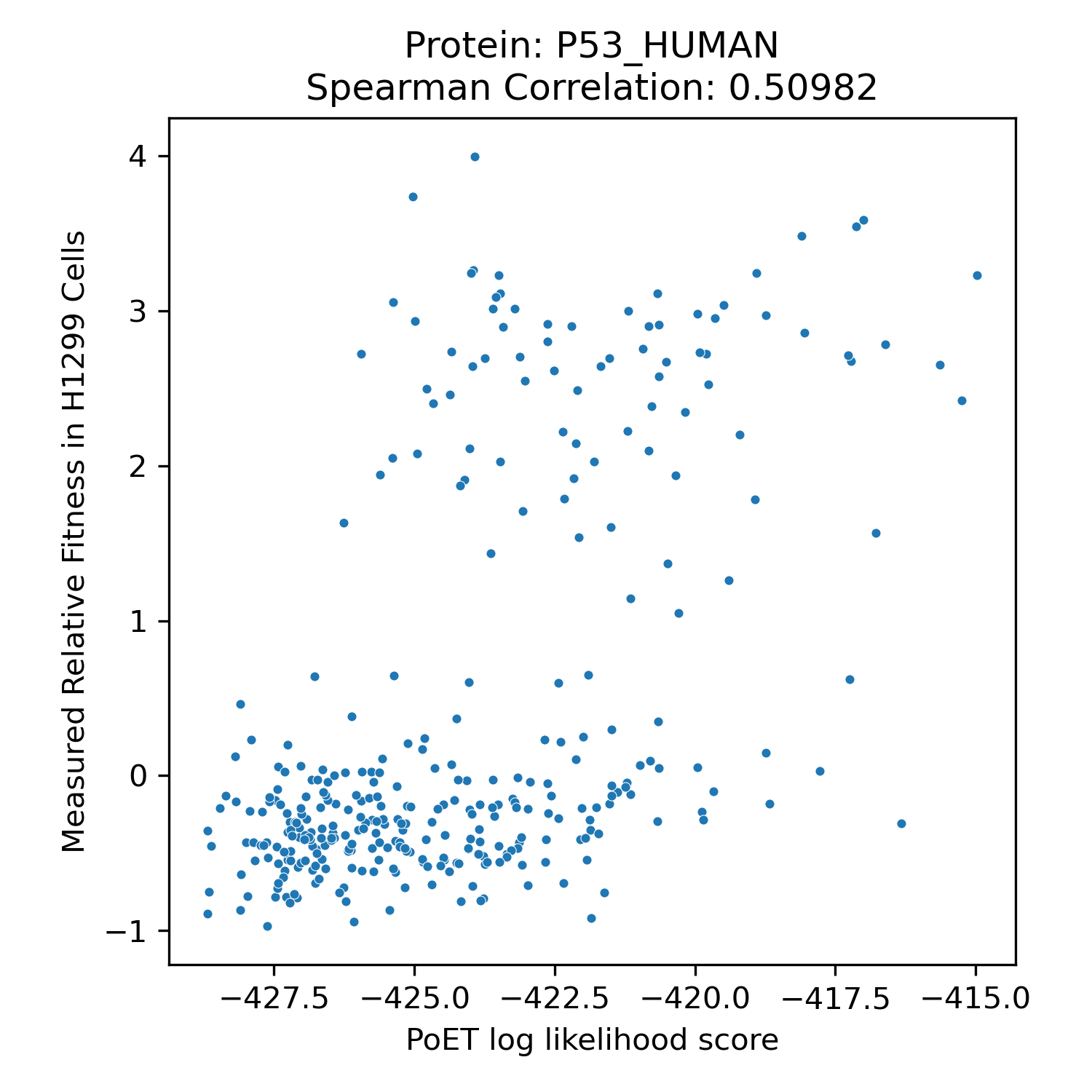
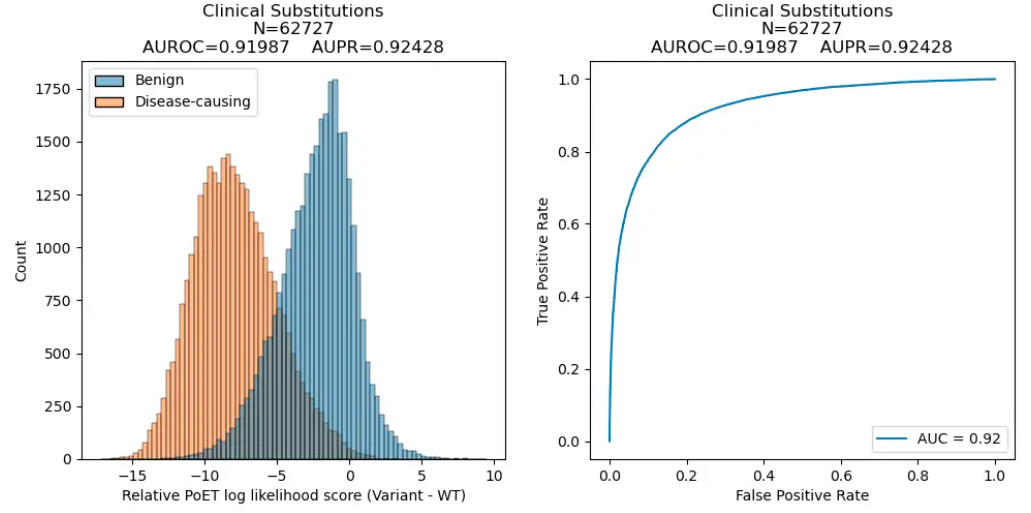
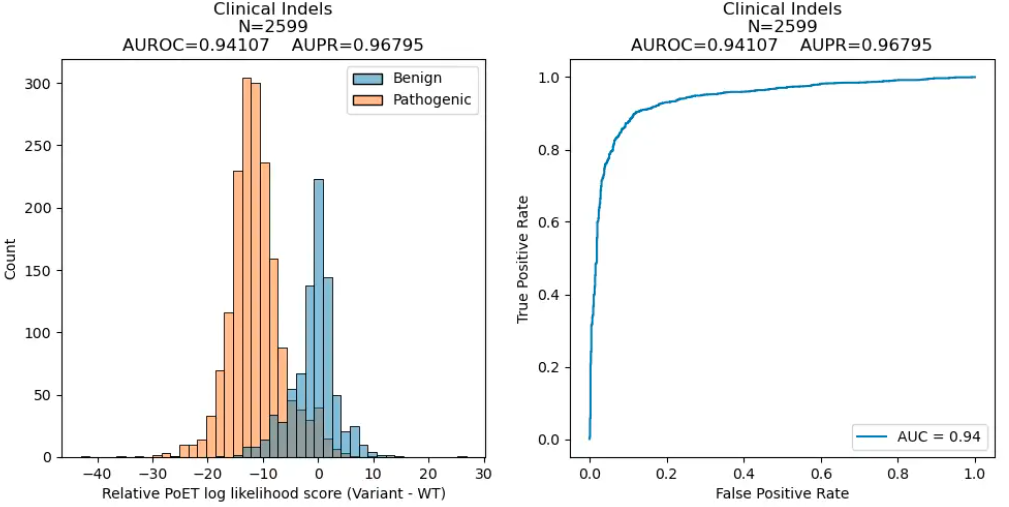
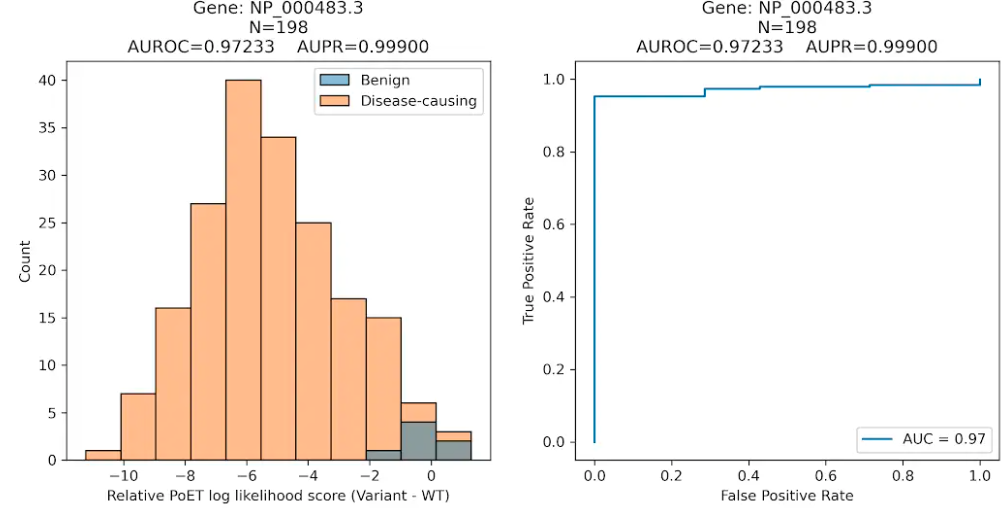
Clinical Variant Prediction
We next evaluated PoET's ability to predict clinical variant effects and distinguish benign from disease-causing mutations using ClinVar data curated in ProteinGym. PoET outperformed other models in both categories, as measured by the Area Under the Receiver Operating Curve (AUROC) metric, which summarizes a model's classification performance at all sensitivity and specificity levels.
Clinical Variant Performance
| Model | Substitutions | Indels |
|---|---|---|
| PoET | 0.924 | 0.941 |
| ESM-1b | 0.892 | N/A |
| ProGen2 | - | 0.847 |
| TranceptEVE | 0.920 | 0.857 |
| EVE | 0.917 | N/A |
| GEMME | 0.919 | N/A |
| PROVEAN | 0.886 | 0.927 |
AUROC of PoET and other commonly used open-source protein language models across substitution and indel variants in ProteinGym's clinical variant dataset.
PoET's superior ability to differentiate disease-causing and benign variants can enhance our understanding of mutation effects in genomes and better prioritize variants from genome-wide association studies or other experiments.
Prompt Engineering for Enhanced Predictions
PoET's predictions can be refined through prompt engineering, where users curate prompts with contextually relevant protein sequences specific to the prediction task. Focused prompts guide PoET's outputs, valuable for engineering proteins with specific functions and contexts, like when engineering specialized enzymes or antibodies.
Users can engineer prompts using sequences based on experimental data or bioinformatically curated sequences from public or proprietary databases. A major advantage of PoET is that it can easily be backed by any database.
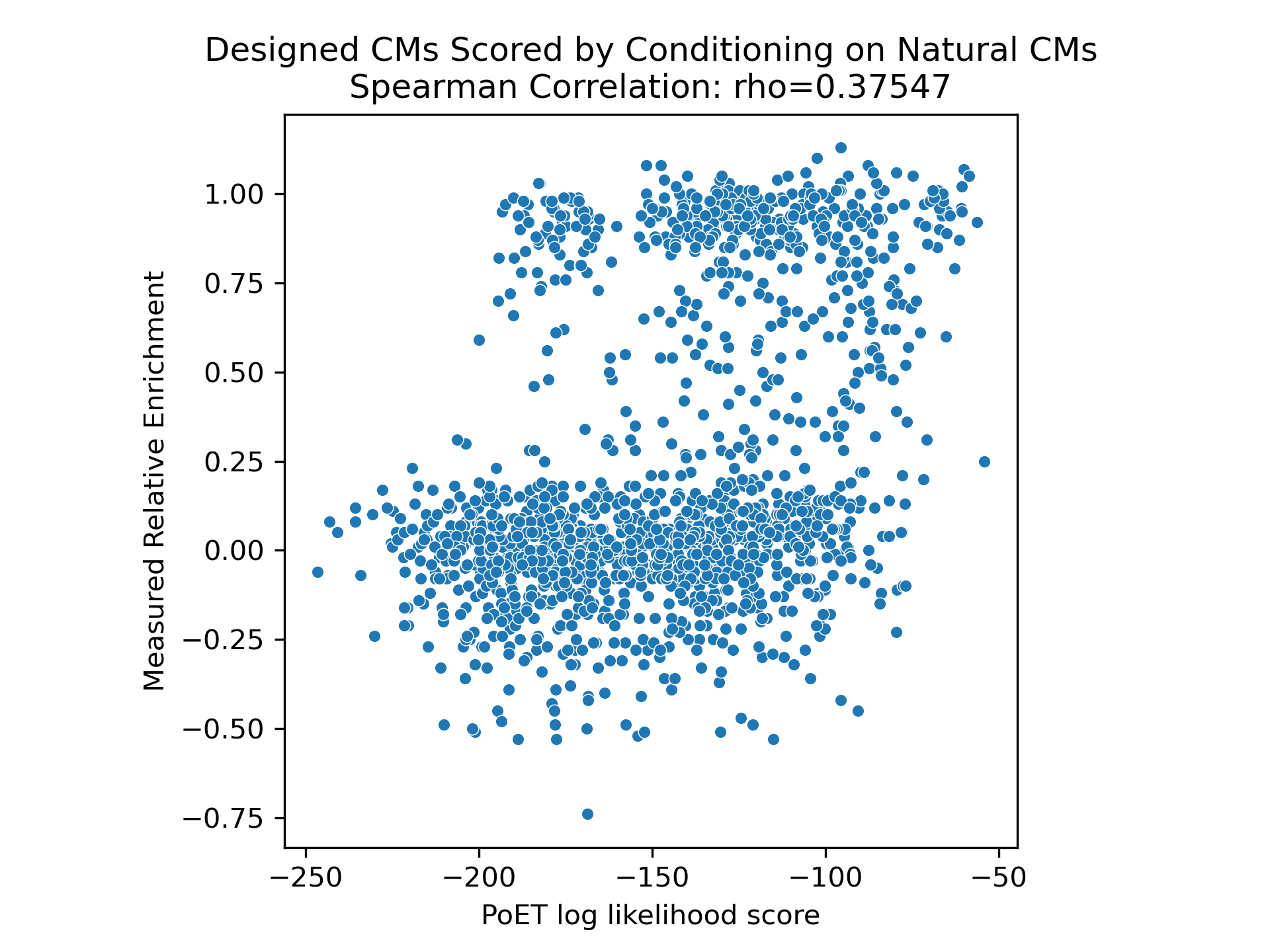
We explored prompt engineering for guiding chorismate mutase generation, an enzyme involved in amino acid biosynthesis. Conditioning on all natural chorismate mutase sequences from a BLAST analysis, PoET performed reasonably well. However, designs significantly improved when PoET was conditioned on functional enzymes with experimentally demonstrated catalytic activity in the cellular environment (Russ et al., 2020).
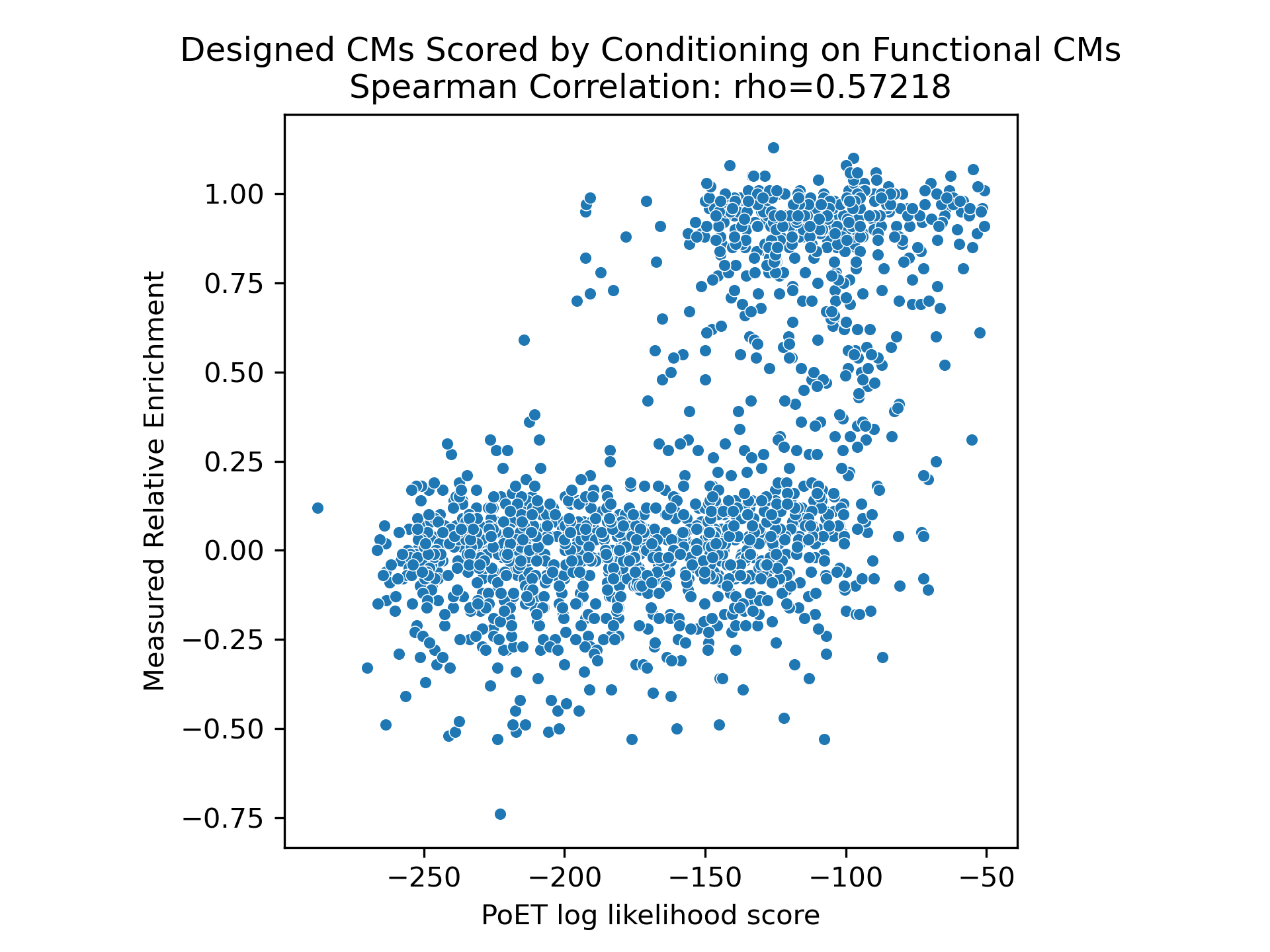
Novel Sequence Generation
Beyond sequence fitness prediction, PoET can generate novel sequences that preserve key constraints inferred from the prompt while introducing new diversity. This enables in silico biopanning and discovery of novel functional sequences. PoET's sequence generation is controllable through homology conditioning, producing sequences with low identity to the original but following the same latent functional and structural distribution.
Visualizing PoET-generated sequences with AlphaFold2 reveals that they preserve the parent sequence's structure despite numerous mutations. This suggests that the generated sequences will fold and demonstrate similar function, despite low sequence identity.
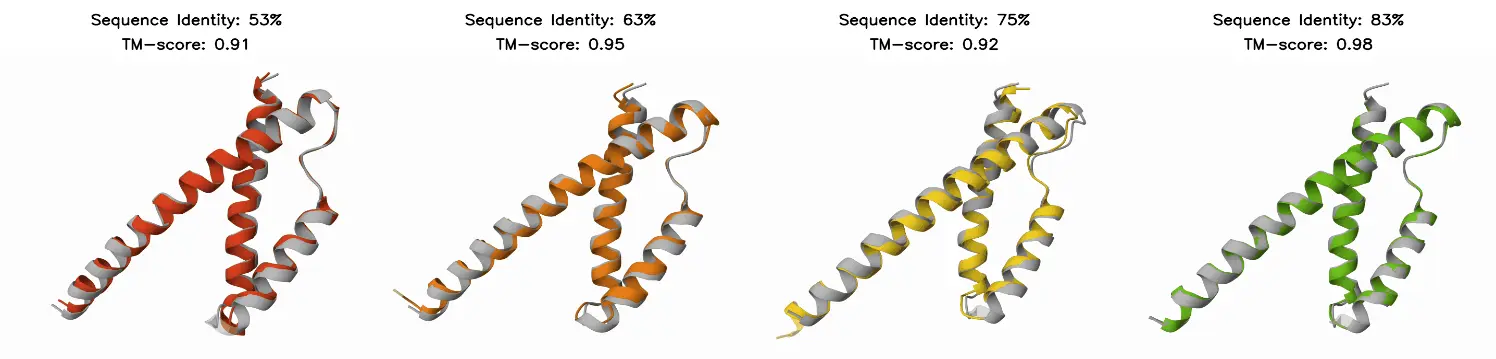
Efficiency and Scalability
PoET is an efficient, quick, and scalable model. It has faster inference speeds and requires less computing power than alternative language models, translating to improved model performance, increased access to accurate predictions, and overall better protein designs. Larger models with more parameters are slower and have decreased computational intensity, while other models like TranceptEVE require hours of pre-training or fine-tuning before they are able to make predictions.
We assessed PoET's efficiency and accuracy with a perplexity evaluation, which measures the ability of a model to accurately predict which amino acid belongs at each position in a protein sequence. The lower the perplexity score, the more accurate the model's predictions. Looking at protein families with few homologs and thus little evolutionary context to base predictions on, PoET outperforms the largest ESM-2 model despite having 75 times fewer parameters. The analysis suggests that matching PoET's performance would take a model with 500B parameters (5 times larger than the largest published protein language model, xTrimoPGLM), making PoET 2500 times more efficient. With its unique ability to provide relevant predictions for proteins with few homologs, PoET offers to expand the space of designable proteins.
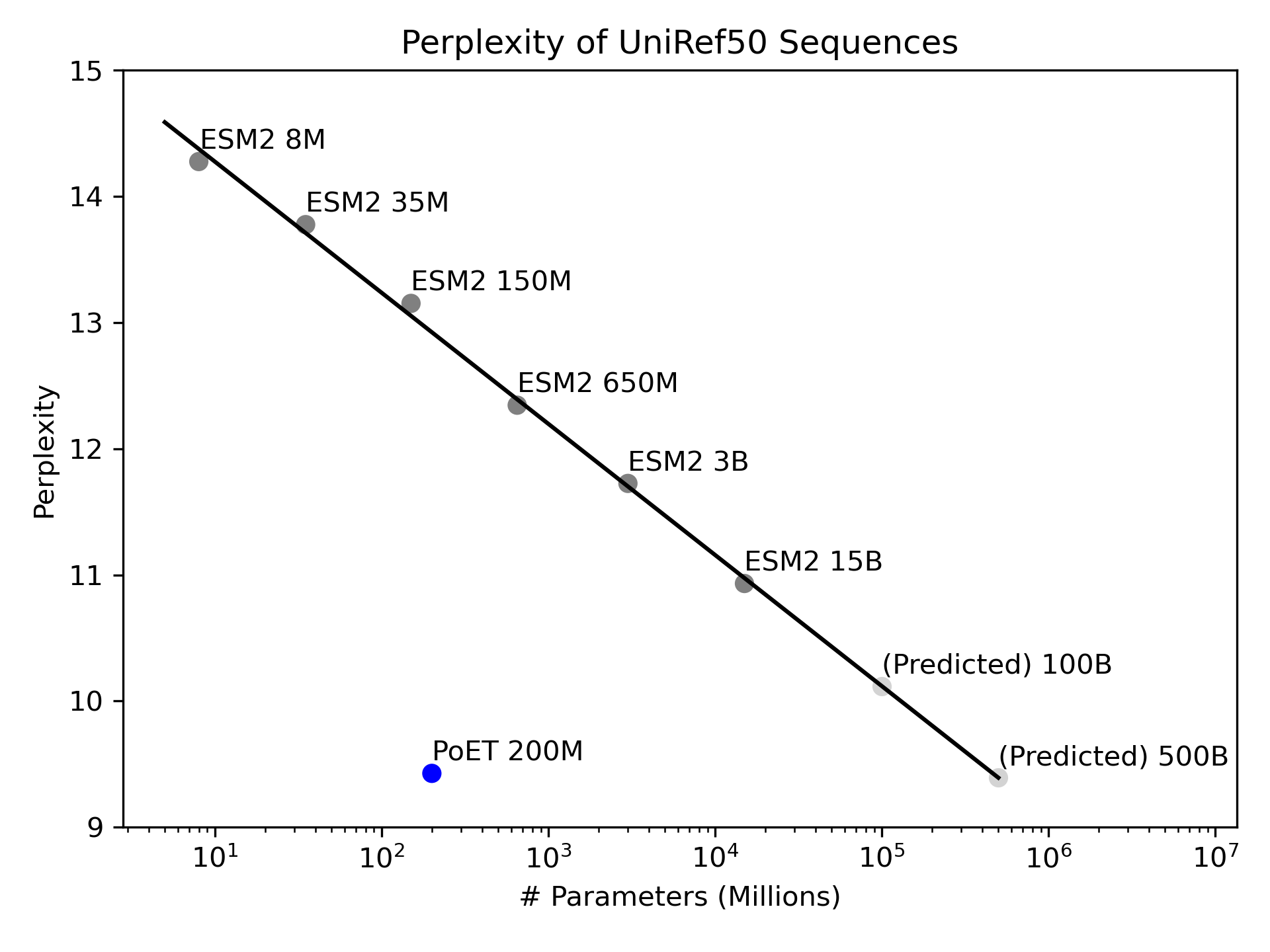
Conclusion and Availability
PoET's unique combination of generalizability, accuracy, controllability, and scale makes it an invaluable tool for guiding zero-shot predictions without prior data collection. Readily available on the OpenProtein.AI platform, PoET enables anyone interested in protein engineering or variant-effect prediction to answer questions like "Which sites should I mutate?", "Which variants should I try?", and "Is this mutation likely to have clinical consequences?".
Sign up for early access to start using PoET in your projects today! You can also learn more about how to use PoET in our docs. For more details, dive into our NeurIPS publication and code on GitHub.
References
Timothy Truong Jr and Tristan Bepler. PoET: A generative model of protein families as sequences-of-sequences. In A. Oh, T. Naumann, A. Globerson, K. Saenko, M. Hardt, and S. Levine, editors, Advances in Neural Information Processing Systems, volume 36, pages 77379–77415. Curran Associates, Inc., 2023. URL
Echave J, Wilke CO. Biophysical Models of Protein Evolution: Understanding the Patterns of Evolutionary Sequence Divergence. Annu Rev Biophys. 2017 May 22;46:85-103. PMID: 28301766. URL
Notin P, Kollasch AW, Ritter D, van Niekerk L, Paul S, Spinner H, Rollins N, Shaw A, Weitzman R, Frazer J, Dias M, Franceschi D, Orenbuch R, Gal Y, Marks DS. ProteinGym: Large-Scale Benchmarks for Protein Design and Fitness Prediction. bioRxiv [Preprint]. 2023 Dec 8:2023. PMID: 38106144. URL
William P. Russ, Matteo Figliuzzi, Christian Stocker, Pierre Barrat-Charlaix, Michael Socolich, Peter Kast, Donald Hilvert, Remi Monasson, Simona Cocco, Martin Weigt, and Rama Ranganathan. An evolution-based model for designing chorismate mutase enzymes. Science, 369 (6502):440–445, 2020. URL