Learning the Grammar of Biology: Why Protein Machine Learning Works
Machine Learning (ML) models have transformed our understanding of protein structure and function. By allowing us to predict complex biophysical properties from sequence alone, they have capabilities that seem unbelievable by the standards of just 10 years ago. Beyond prediction, we can now generate novel sequences with properties that we desire. There is no magic in these models. Nor is their success a function of accumulating more labeled data alone. Rather, they inherit a decades-long tradition of understanding how amino acid sequences shape protein structure and function and reflect underlying evolutionary patterns. Translating these biological rules into modern ML models has given us unforeseen powers, by enabling us to learn the patterns inscribed by molecular evolution.
Proteins are the foundation of molecular biology. From 20 amino acids, encoded in DNA, proteins fold into diverse shapes and forms, performing most of the functional roles in molecular biology. Biochemically, it is the structure and chemical properties of the exposed surface of a protein that determines its function. By folding rapidly and with extremely high fidelity, proteins take on roles ranging from structural channels, to molecular motors, to ion channels, signal transducers, and more, within the crowded environment of the cell.
It is widely believed that protein structure determination is essential to our ability to understand and manipulate the molecular functions of proteins. Mapping structure experimentally is limited by cost and scale, creating the need for computational structure prediction methods. Historically, approaches for structure prediction were mostly physics-based, attempting to model interactions between neighboring amino acids alongside the polymer properties of the peptide chain itself. Yet despite decades of research into these first-principle approaches, protein structure and function prediction have remained intractable until recently.
The Non-Random Nature of Protein Sequences
The distribution of proteins in the natural world is non-random. Patterns in these sequences reflect the functional and structural constraints imposed by natural selection.
Another approach considers the protein sequence alone. While structure conveys function, it is the underlying DNA, encoding protein sequences, that evolves. Billions of years of adaptive radiation have led to the vast variety of proteins we observe in the natural world. Without purifying selection guiding towards a set of general "rules," neutral evolution would diversify sequences independently in each organism. But, the distribution of proteins in the natural world is non-random. Patterns in these sequences reflect the functional and structural constraints imposed by natural selection. We can intuit what these constraints are from our knowledge of biology: non-immunogenicity, stability, lack of aggregation, conservation of function, etc. in the physiological conditions of the organism.
Evolutionary Sequence Modeling: A Data-Driven Approach
Evolutionary sequence modeling, as it has been performed for half a century or more, seeks to extract this information from natural sequences. Identify similar sequences either within or across organisms. Define a protein family. Align those sequences and identify the conserved amino acids and positions within them and their coevolutionary dependencies on each other. Relate these changes to structural properties (e.g., where they bind) or functional ones (who they bind to).
These methods enable a fundamentally different, data-driven approach to structure and function prediction. They have also grown in complexity and power over time as the volume of sequencing data and computing power available have increased, progressing from basic alignment methods to position-specific scoring matrices (PSSMs) to Hidden Markov Models (HMMs) and Potts models. This was made possible due to decades of accumulated protein sequences in UniProt and accompanying structure and function datasets such as the Protein Data Bank (PDB). While powerful on the scale of a single protein, these models are fundamentally unable to scale to the vast diversity of sequenced proteins. A more general tool is needed.
The Statistical Nature of Biological "Grammar"
Biological "grammar" is composed of statistical patterns. Certain combinations of residues tend to fold certain ways, leading to stereotyped secondary structures, such as alpha helices and beta sheets. Functional domains are often not strictly stereotyped, but rather reflect diverse and distinct combinations of amino acids in sequence that convey the same chemical properties. In gene expression, the search for stereotyped binding motifs has been unable to fully explain the variety of regulatory patterns and states we find. Similarly, innate immunity in eukaryotes relies on recognition of patterns of residues likely to originate from pathogens, both definite and statistical. Biology lies in statistical patterns not stereotyped motifs.
Deep Language Models: Finding Patterns in Biology
Deep Language Models are designed to find exactly these patterns. Originally developed in the context of Natural Language Processing (NLP), these models infer grammar and semantics from text without expectations of their organization, beyond the sequential ordering of words. Thus, we can build language models that can translate between languages with completely opposite word and clause orderings (e.g., English and Japanese). Mathematically, they are generative models: they generate sequences word by word (or token by token), based on probabilistically choosing the most likely next element ("generative") given what has been seen before. Their generality allows them to be trained on an entire corpus of text and applied broadly across it. Unlike an HMM, we do not have to specify a sequence family that a model is used for. Applying Deep Language Models to biological sequences is a natural extension. We replace word tokens with amino acid ones and train on sequence databases like UniProt.
By modeling sequences of arbitrary length and function, we can begin to learn statistical properties of proteins outside of specific domains and families. In mathematical terms, we are learning the manifold of natural protein sequences. We should expect the "grammar" learned from these proteins to reflect the statistical properties we discovered previously: secondary structures like alpha helices and beta sheets, transmembrane domains, and more. A dangerous sequence is one that is unlikely under the model. Evolution is very unlikely to generate it, suggesting that it is unfit for its physiology and/or likely toxic.
Protein Design as Language Generation
Just as we can generate text about a subject and iteratively improve it, we can design novel protein sequences that are natural to an organism or for a function.
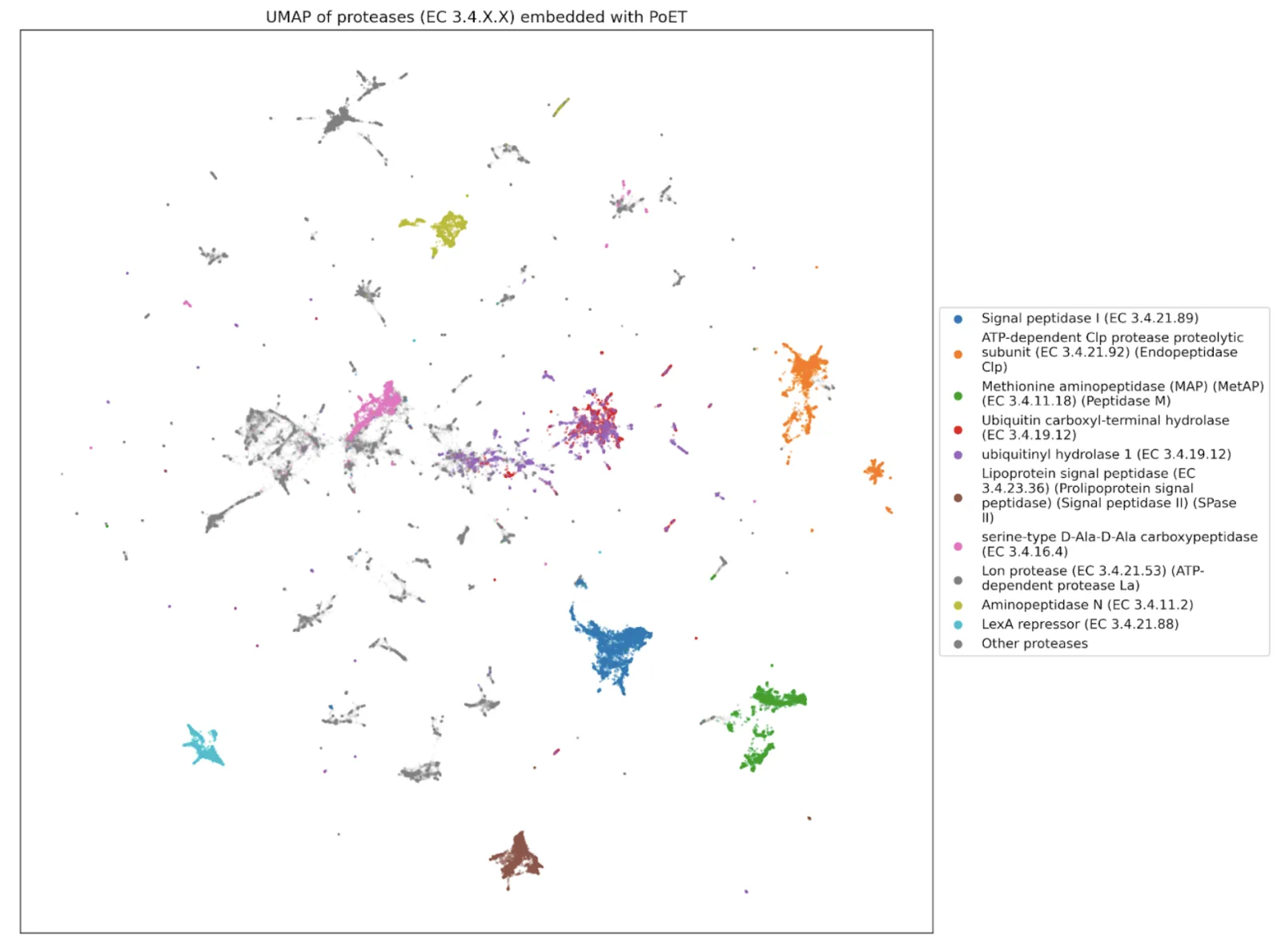
We expect, too, to identify new properties that must exist but could not have been found by previous methods. Just as our innate immune system can recognize likely pathogenic sequences based on local sequence properties, so too should a language model. This is exactly what we find with modern language models.
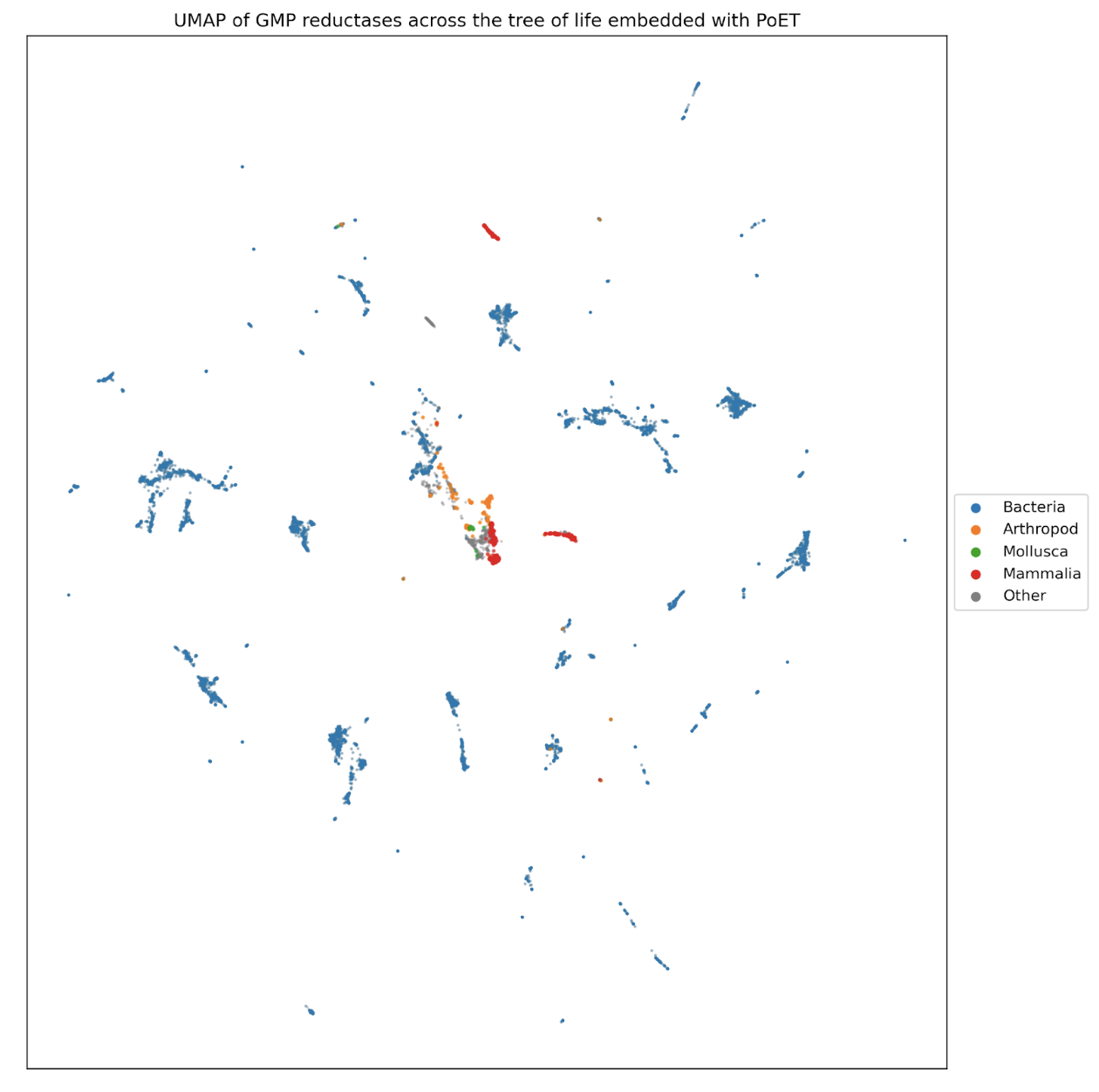
From this vantage, engineering proteins mirrors our everyday interactions with language: generating and editing sequences of amino acids, just as we would sentences. Just as we can generate text about a subject and iteratively improve it, we can design novel protein sequences that are natural to an organism or for a function. For instance, increasing the stability of an enzyme at high temperatures, or optimizing binding to a set of molecules while avoiding binding to certain others. We can also translate between systems, just as with language. If we want to increase the expressibility of a human protein in yeast, we can generate sequences likely to be found in yeast conditioned on similarity to human proteins. Similarly, if we want to humanize an antibody developed in mice, we can generate framework regions conditioned on human antibody sequences.
Looking Forward
At OpenProtein.AI, we've been developing protein language models that capture these complex statistical patterns in new ways, enabling us to push the envelope of structure and function prediction and protein design. In upcoming posts, we will discuss in greater detail how OpenProtein.AI's language models relate to previous computational tools in protein design such as HMMs, Statistical Coupling Analysis and more, as well as how we can exploit their power to design new ways of optimizing proteins better than a scientist or a computer alone.